2023.10.03
HyenaDNA:
LLMでゲノム解析してみた(入門編)
はじめに
こんにちは、グループ研究開発本部・AI研究室のT.I.です。前回のBlogでは、Hyenaという新しい機械学習モデルをDNA配列の解析に応用するという HyenaDNA という研究を簡単に紹介してみました。 HyenaDNAは、GitHub (https://github.com/HazyResearch/hyena-dna) にて実装が公開されていますので、今回のBlogでは、実践編として公開されているコードを元にHyenaDNAを実際に動かしてみたいと思います。
Genomic Benchmarks
前回のBlogで紹介したように、HyenaDNAではいくつかのベンチマークで評価されています。 今回は、そのなかの1つであるGenomic Benchmarks( https://github.com/ML-Bioinfo-CEITEC/genomic_benchmarks)を利用してみます。 Genomic Benchmarksを利用をするためには、以下のコマンドでインストールします。
$ pip install --quiet genomic-benchmarks
利用可能なデータセットは、以下のコマンドで確認できます。
from genomic_benchmarks.data_check import list_datasets list_datasets()
- Mouse Enhancers
- Coding vs Intergenomic
- Human vs Worm
- Human Enhancers Cohn
- Human Enhancers Ensembl
- Human Regulatory
- Human Nontata Promoters
- Human OCR Ensembl
- Drosophila enhancers
ここで、最初の8つのタスクについて、HyenaDNAでは性能比較を行っています。ここで最後のDrosophila(ショウジョウバエ) Enhancersは、 HyenaDNAでは使用されていませんが、おそらくヒトとは関係ないので評価していないのかと思われます。 なお、Enhancerとは転写因子と呼ばれるタンパク質が結合し、その上流(下流)の遺伝子の発現を調節するDNA配列のことです。 GenomicBenchmarkでの結果は以下の表のように、HyenaDNAの性能がCNNを大きく上回り、8個中7個のタスクでTransformerよりも高性能です。
| Dataset | CNN| | Transformer| | HyenaDNA |
|---|---|---|---|
| Mouse Enhancers | 69.0 | 80.1 | 84.3(+15.3) |
| Coding vs Intergenomic | 87.6 | 88.8 | 87.6(+3.5) |
| Human vs Worm | 93.0 | 95.6 | 96.5(+3.5) |
| Human Enhancers Cohn | 69.5 | 70.5 | 73.8(+4.3) |
| Human Enhancers Ensembl | 68.9 | 83.5 | 89.2(+20.3) |
| Human Regulatory | 93.3 | 91.5 | 93.8(+0.5) |
| Human Nontata Promoters | 84.6 | 87.7 | 96.6(+12) |
| Human OCR Ensembl | 68.0 | 73.0 | 80.9(+12.9) |
HyenaDNAの論文の表4.1より作成したGenomic Benchmarkの性能(Top-1 Accuracy)比較結果
データセットの詳細は、
import genomic_benchmarks
genomic_benchmarks.data_check.info('drosophila_enhancers_stark', version=0)
で確認できます。
Dataset `drosophila_enhancers_stark` has 2 classes: negative, positive. The length of genomic intervals ranges from 236 to 3237, with average 2118.1238067688746 and median 2142.0. Totally 6914 sequences have been found, 5184 for training and 1730 for testing.
このデータセットでは、positiveとnegativeの2クラス分類を行うことになります。入力長としては、236から3237の間の長さの配列が与えられます。データセットのダウンロードは以下の通りです。
genomic_benchmarks.loc2seq.download_dataset('drosophila_enhancers_stark', version=0, dest_path='.')
個別のファイルは、以下のようなフォルダ構成になっています。
$ tree -L 2 drosophila_enhancers_stark
drosophila_enhancers_stark
├── test
│ ├── negative
│ └── positive
└── train
├── negative
└── positive
データはそれぞれ、アデニン(A)、シトシン(C)、グアニン(G)、チミン(T)の文字列で表現されています。例えば、以下のようなファイルがあります。
$ cat drosophila_enhancers_stark/test/negative/999.txt CCGCCGACGGCCAGCAGAACCGGACGCAGTACCAGCCGCCTTGACATCAGCGACATGGAATCCATTTATGGGTACCTAAAGCCCCGCAAGCCGCGCAGCCTCAGCCTGAAGAAAATCCAAATACTCGACTACTAAACGGACACGTGAATTTCTATGGGCTTGCTCTAAATCGAAACGAGGTTATATGGCCCAGACATCGATATTAGTTATTGTTTTTTGTTGCCCTATCCCCTTGGAGCGGATGATGGGTGGCTGTACATACATATATGCTATATTATACCCACAAACGCATCTCGGTTACCTCCTTGCCCGGTGAACAGAACCACTAAACTGTACGATACTTTTACTAACATAGCACCTTGGACTCCTCTAGCCTGGACAGTCACGAAACGCAAATGTACATACACCAAGTGGGAGTGGGTTACTGATTTTAGAGGTAGTCCAAAGTTCAAACGAGTGGGTTGTAATCTGTGGAATAATACACTTACATTACATTATATCCCAAAAAAGAGATAATGCAATTAAACAGATAATACCTTCACCAGATTACAACCAAGTACCCGCTTGGGTCTACCAATAGAATCCACCCACCCCAGCACACATAATTATCCCGACAATTGTACACGCACCCGATTTTTAACCACGGATTTCTGTGCTTCTTGTAGATTCGGAGCTTGTTGAATCACCCTACAATCACATCTACGGACGCCATTTACCGCTGCCAACGCGCGGCTACGTGCCCACGCCACGCATGTTCATCGGGGAATGGGACTGAGCGTAGTTCACACCGCACCTGGGCGGGATTTTAGCCGGATTTTGAGCGGCCAAGGCACAAAACACACCACAACACTTGCACATCCCGGCGTGAGAGGGAACGACGCCTACCGTGCAGAGCTAATTTAAGTCAACCGGCTGCACACAGCTTGAAATATTATTTACATTATATTAATTTAGCAAAATATTATGGTAAAATAATATTAGGCTAGGCATAAGCAGCAACAAAAAAAACAAGAGTAATCGCAAACCATTGTAACATTTAGTGCCGTGATTTTTCGCGCTTTTCCCCTAGCAATTTTGAGATTACGCACTTGAGCCAATTTTGTTTACCCTGTTGTACGAAATGAATGAATAGCCATTACATCACGAGTTCAGTTAAGCTTTTAAGTATATTGTAAATTTCTCTTGAGCAGCCGTCAACTCAACTGGCCTGTTTGCGTATTTACTTGATAAATGTTCTTAACTACGAATTTTTCTAGTAAAAACTCAAACAAGCGTTGTCTAACAGGCTGCTGTTTTTTATTTTATATCTGTAAAATTCTGTAATATTCTGTATTGTGTATGCAACGTAATAAAAATAATCTTTTAAAACAATCCGTTTAATTTTGGTCTTCTAAAATGAAAACTTTTTTCATTGCTTTAAACTAAAGATTTGAATTGCTCACTATACATGGCATTCCTGGTAATTACATGTAGTAATCGGGATTCCAATCTGGTGGGCGCATATGATCTGGATTGATTCCACCCTGTCCCGGCCTATTCGGGTTCAGGGGATTGAACGGGTCGAAGCGGGGCACGGGTCCCGGGCCCATATTTGGGCGTGATGGAAAGCTGAACAGGTTACCATGACCACCGCGACCCAGTGGATCGAGATCCCCGCGTCCAACGTCCGGAAATCCAAAAGGGCGAGGCTCGAATGCACTTGGAATGAAGGATCTAAACGAGGGGAAACGATTAACGAGTGTATAAAAAACTATCCACAAGTGTTCTTACCCGCCACGCCTTGGCTCGCCGATGCGCAAGGGATCTGGATCCGATCCAATGGGGCGTGGTGAGTTGGTCGTCTGCGTGGTAACTTCGCGCGAGTTTCCCGTGAATACAGGGTCTAGAAGCTCCCTACGGTAGCGTTCAACAATCTCTGAGGCACTCGGCATTATGGTGGTGATGCCTCCCTTGACCTCAGGTACCAGGGTCTCTGGTTCTACGCAGATGTTGGATACCTTCTTAGTGTTGATGTCCAACAGGTTGATCAGCAGGGAGCCCTCGGTGATGTGGCCCAGCAGCAAATACAGCATCTTATCGTGCACGTAGCGCAGCGAGTACTTGGTATCGTCATCATTCCAGCTGTCCGGCAGCAGCTCACTGCCCTCCTCCTCCGGCAAGGTTTTCTATGGAATTCCAGATGATTTCACTAATATGCGTGGCCATAATCATACTCACATCATCGCCAACGCCAACG
HyenaDNAを(ちょっと)動かしてみる
HyenaDNAはPyTorchで実装されています。具体的なインストール方法については、公式レポジトリで解説がありますが、今回は簡単のため1つのファイルだけでHyenaDNAのモデルが実装されているstandalone_hyenadna.py (https://github.com/HazyResearch/hyena-dna/blob/main/standalone_hyenadna.py) を実行してみます。なお、huggingface.py (https://github.com/HazyResearch/hyena-dna/blob/main/huggingface.py) を利用すると事前学習済みモデルを利用できます。これらはヒトのレファレンスゲノム配列(hg38)を使用して、LLMのように次の塩基の予測(Next Token Prediction)で事前学習されています。以下が利用できるモデルの一覧です。モデル名の後にあるのはそれぞれ事前学習、ファインチューニング、推論に利用するのに必要なGPUのリソースです。
- tiny-1k (T4 16GB, T4 16GB, T4 16GB)
- tiny-1k-d256 (T4 16GB, T4 16GB, T4 16GB)
- small-32k (A100 40GB, T4 16GB, T4 16GB)
- medium-160k (A100 40GB, A100 40GB, T4 16GB)
- medium-450k (A100 40GB, A100 40GB, T4 16GB)
- large-1m (A100 80GB, A100 80GB, A100 40GB)
特に、大型モデルの学習時は前回のBlogで解説したように短い配列の学習から始めて、段階的に長い配列の学習をしています。
HyenaDNAの事前学習のアニメーション(前回Blogより)
まず、最初に必要なlibraryをインストールします。
$ pip install --quiet einops torch torchvision transformers OmegaConf
そして、実行に必要なソースコードもダウンロードしておきます。
$ wget https://raw.githubusercontent.com/HazyResearch/hyena-dna/main/standalone_hyenadna.py $ wget https://raw.githubusercontent.com/HazyResearch/hyena-dna/main/huggingface.py
HyenaDNAのモデルを作成するには以下のようにします。(なお、以下のコードなどは公式リポジトリで公開されているものを元に作成してあります。)
import torch
from standalone_hyenadna import HyenaDNAModel
use_head = True
n_classes = 2
backbone_cfg = {
"d_model": 128,
"n_layer": 2,
"d_inner": 512,
"vocab_size": 12,
"resid_dropout": 0.0,
"embed_dropout": 0.1,
"fused_mlp": False,
"fused_dropout_add_ln": True,
"residual_in_fp32": True,
"pad_vocab_size_multiple": 8,
"return_hidden_state": True,
"layer": {
"_name_": "hyena",
"emb_dim": 5,
"filter_order": 64,
"local_order": 3,
"l_max": 1026,
"modulate": True,
"w": 10,
"lr": 6e-4,
"wd": 0.0,
"lr_pos_emb": 0.0
}
}
device = 'cuda' if torch.cuda.is_available() else 'cpu'
print("Using device:", device)
model = HyenaDNAModel(
**backbone_cfg, use_head=use_head, n_classes=n_classes
)
事前学習済みのモデルを利用する場合は、以下のようにします。
import torch
from huggingface import HyenaDNAPreTrainedModel
pretrained_model_name = 'hyenadna-tiny-1k-seqlen'
use_head = True
n_classes = 2
backbone_cfg = None
device = 'cuda' if torch.cuda.is_available() else 'cpu'
print("Using device:", device)
model = HyenaDNAPreTrainedModel.from_pretrained(
'./checkpoints',
pretrained_model_name,
download=True,
config=backbone_cfg,
device=device,
use_head=use_head,
n_classes=n_classes,
)
Colaboratoryで公開されているサンプル・コードからGenomic Benchmark Datasetを利用する際に必要なモジュールを抜粋すると以下の箇所になります。
import torch
from random import random
import numpy as np
from pathlib import Path
from torch.utils.data import DataLoader
from genomic_benchmarks.loc2seq import download_dataset
from genomic_benchmarks.data_check import is_downloaded
# helper functions
def exists(val):
return val is not None
def coin_flip():
return random() > 0.5
string_complement_map = {'A': 'T', 'C': 'G', 'G': 'C', 'T': 'A', 'a': 't', 'c': 'g', 'g': 'c', 't': 'a'}
# augmentation
def string_reverse_complement(seq):
rev_comp = ''
for base in seq[::-1]:
if base in string_complement_map:
rev_comp += string_complement_map[base]
# if bp not complement map, use the same bp
else:
rev_comp += base
return rev_comp
class GenomicBenchmarkDataset(torch.utils.data.Dataset):
'''
Loop thru bed file, retrieve (chr, start, end), query fasta file for sequence.
Returns a generator that retrieves the sequence.
Genomic Benchmarks Dataset, from:
https://github.com/ML-Bioinfo-CEITEC/genomic_benchmarks
'''
def __init__(
self,
split,
max_length,
dataset_name='human_enhancers_cohn',
d_output=2, # default binary classification
dest_path="/content", # default for colab
tokenizer=None,
tokenizer_name=None,
use_padding=None,
add_eos=False,
rc_aug=False,
return_augs=False,
):
self.max_length = max_length
self.use_padding = use_padding
self.tokenizer_name = tokenizer_name
self.tokenizer = tokenizer
self.return_augs = return_augs
self.add_eos = add_eos
self.d_output = d_output # needed for decoder to grab
self.rc_aug = rc_aug
if not is_downloaded(dataset_name, cache_path=dest_path):
print("downloading {} to {}".format(dataset_name, dest_path))
download_dataset(dataset_name, version=0, dest_path=dest_path)
else:
print("already downloaded {}-{}".format(split, dataset_name))
# use Path object
base_path = Path(dest_path) / dataset_name / split
self.all_paths = []
self.all_labels = []
label_mapper = {}
for i, x in enumerate(base_path.iterdir()):
label_mapper[x.stem] = i
for label_type in label_mapper.keys():
for x in (base_path / label_type).iterdir():
self.all_paths.append(x)
self.all_labels.append(label_mapper[label_type])
def __len__(self):
return len(self.all_paths)
def __getitem__(self, idx):
txt_path = self.all_paths[idx]
with open(txt_path, "r") as f:
content = f.read()
x = content
y = self.all_labels[idx]
# apply rc_aug here if using
if self.rc_aug and coin_flip():
x = string_reverse_complement(x)
seq = self.tokenizer(x,
add_special_tokens=False,
padding="max_length" if self.use_padding else None,
max_length=self.max_length,
truncation=True,
) # add cls and eos token (+2)
seq = seq["input_ids"] # get input_ids
# need to handle eos here
if self.add_eos:
# append list seems to be faster than append tensor
seq.append(self.tokenizer.sep_token_id)
# convert to tensor
seq = torch.LongTensor(seq)
# need to wrap in list
target = torch.LongTensor([y])
return seq, target
モデルの訓練、テストについては以下の通りです。
def train(model, device, train_loader, optimizer, epoch, loss_fn, log_interval=10):
"""Training loop."""
model.train()
for batch_idx, (data, target) in enumerate(train_loader):
data, target = data.to(device), target.to(device)
optimizer.zero_grad()
output = model(data)
loss = loss_fn(output, target.squeeze())
loss.backward()
optimizer.step()
if batch_idx % log_interval == 0:
print('Train Epoch: {} [{}/{} ({:.0f}%)]\tLoss: {:.6f}'.format(
epoch, batch_idx * len(data), len(train_loader.dataset),
1. * batch_idx / len(train_loader), loss.item()))
def test(model, device, test_loader, loss_fn):
"""Test loop."""
model.eval()
test_loss = 0
correct = 0
with torch.no_grad():
for data, target in test_loader:
data, target = data.to(device), target.to(device)
output = model(data)
test_loss += loss_fn(output, target.squeeze()).item() # sum up batch loss
pred = output.argmax(dim=1, keepdim=True) # get the index of the max log-probability
correct += pred.eq(target.view_as(pred)).sum().item()
test_loss /= len(test_loader.dataset)
print('\nTest set: Average loss: {:.4f}, Accuracy: {}/{} ({:.2f}%)\n'.format(
test_loss, correct, len(test_loader.dataset),
100. * correct / len(test_loader.dataset)))
Downloadしたデータを訓練・テストに分けてモデルで実行できるようにTokenizerを作成し、データセットを作成します。 ここでデータセットとしては、Human Enhancers Cohn(Enhancer Identification using Transfer and Adversarial Deep Learning of DNA Sequences)を利用します。これは、総数27,791の長さ500の塩基配列のデータセットで、positiveとnegativeの2クラス分類を行うものです。
from standalone_hyenadna import CharacterTokenizer
max_length = 500
tokenizer = CharacterTokenizer(
characters=['A', 'C', 'G', 'T', 'N'], # add DNA characters, N is uncertain
model_max_length=max_length + 2, # to account for special tokens, like EOS
add_special_tokens=False, # we handle special tokens elsewhere
padding_side='left', # since HyenaDNA is causal, we pad on the left
)
use_padding = True
dataset_name = 'human_enhancers_cohn'
batch_size = 256
rc_aug = True # reverse complement augmentation
add_eos = False # add end of sentence token
ds_train = GenomicBenchmarkDataset(
max_length = max_length,
use_padding = use_padding,
split = 'train',
tokenizer=tokenizer,
dataset_name=dataset_name,
rc_aug=rc_aug,
add_eos=add_eos,
)
ds_test = GenomicBenchmarkDataset(
max_length = max_length,
use_padding = use_padding,
split = 'test',
tokenizer=tokenizer,
dataset_name=dataset_name,
rc_aug=rc_aug,
add_eos=add_eos,
)
train_loader = DataLoader(ds_train, batch_size=batch_size, shuffle=True)
test_loader = DataLoader(ds_test, batch_size=batch_size, shuffle=False)
損失関数としてCross Entropy Lossを利用し、optimizerを作成します。
import torch.optim as optim
import torch.nn as nn
# loss function
loss_fn = nn.CrossEntropyLoss()
learning_rate = 6e-4 # good default for Hyena
weight_decay = 0.1
# create optimizer
optimizer = optim.AdamW(model.parameters(), lr=learning_rate, weight_decay=weight_decay)
さて、ようやくモデルの訓練を行います。
model.to(device)
num_epochs = 10
for epoch in range(num_epochs):
train(model, device, train_loader, optimizer, epoch, loss_fn)
test(model, device, test_loader, loss_fn)
optimizer.step()
以上を実行すると以下のようにHyenaDNAのモデルの学習とテストが進んでいきます。
Train Epoch: 0 [0/20843 (0%)] Loss: 0.690840 Train Epoch: 0 [2560/20843 (12%)] Loss: 0.569274 Train Epoch: 0 [5120/20843 (24%)] Loss: 0.549916 Train Epoch: 0 [7680/20843 (37%)] Loss: 0.591534 Train Epoch: 0 [10240/20843 (49%)] Loss: 0.565957 Train Epoch: 0 [12800/20843 (61%)] Loss: 0.517197 Train Epoch: 0 [15360/20843 (73%)] Loss: 0.614621 Train Epoch: 0 [17920/20843 (85%)] Loss: 0.516811 Train Epoch: 0 [20480/20843 (98%)] Loss: 0.516065 Test set: Average loss: 0.0022, Accuracy: 4967/6948 (71.49%) Train Epoch: 1 [0/20843 (0%)] Loss: 0.530085 Train Epoch: 1 [2560/20843 (12%)] Loss: 0.528274 Train Epoch: 1 [5120/20843 (24%)] Loss: 0.527972 Train Epoch: 1 [7680/20843 (37%)] Loss: 0.527023 Train Epoch: 1 [10240/20843 (49%)] Loss: 0.521246 Train Epoch: 1 [12800/20843 (61%)] Loss: 0.543012 Train Epoch: 1 [15360/20843 (73%)] Loss: 0.540115 Train Epoch: 1 [17920/20843 (85%)] Loss: 0.544496 Train Epoch: 1 [20480/20843 (98%)] Loss: 0.495625 Test set: Average loss: 0.0021, Accuracy: 5084/6948 (73.17%) ...
試しに適当な配列を入力して遊んでみます。
import numpy as np
def inference_example(model, device, max_length=100):
sequence = ''.join(np.random.choice(['A', 'C', 'T', 'G'], max_length))
print('# input sequence:', sequence)
tok_seq = tokenizer(sequence)
tok_seq = tok_seq["input_ids"] # grab ids
# place on device, convert to tensor
tok_seq = torch.LongTensor(tok_seq).unsqueeze(0) # unsqueeze for batch dim
tok_seq = tok_seq.to(device)
# prep model and forward
model.to(device)
model.eval()
with torch.inference_mode():
output = model(tok_seq)
pred = output.argmax(dim=1).item()
print(f'# prediction:', pred)
for i in range(10):
inference_example(model, device)
なんかそれっぽい、結果が出てきましたね(まあ、ランダムな配列なんで意味はないですが)。
# input sequence: GGCTTCGAGCAGTGAACGCACGTGGTTTCAGGAGAGGCCGGGAGTTTTCTTTAGGCATCAGAGCCATCGGACAACGTGGTTCGCCCTAATAAATCCGCTC
# prediction: 0
# input sequence: GCACATTGACCGGGGTACGTGTCGTTATGACCTGTGTGAATCTTGGGGTCGAATATGCCGTGTGATTGACGCCGCTTGATTTATCCCGTGTCGCTCCGAA
# prediction: 0
# input sequence: TGGTTCTTATCACTCTCAAAGAAGGTCTCCCCAGACGGCACGTTTTGACCTGACGGGTGTCTGACTGTCCTTAGGAACAAAGATCGTGTGTATGCTGGGG
# prediction: 0
# input sequence: CTAGAACCAGAACCGGGGACAGAACGCCGTTCACTAGACGCGGGGTTTTGGTCGCGCTCCCGCCGACGTGAAAGTCTATCTTCTCCCATAGCCTTCCAAT
# prediction: 0
# input sequence: CTCAATCCGGAGCCGTCATATTCTATGTGCTCCTCCTTACGACCTATTTTATATTACATTGTTGGGGATGCATGCGTTCTCTCGTCGTGCTGTTATCTGA
# prediction: 0
# input sequence: ATCGGATGGTATACCAGCGGTTGCACAAACAACAAACGAGCACCATCTTGTTTCCCTCATGACAGCTCTACGATGGCATCTGAGTCGATATCACTCGGGG
# prediction: 1
# input sequence: AATTATAAGGCAAGTAGCGGGCGCGCATAGGCTTCAACCACACTTGAAAGAGTCAGTTTGTTACGCAAACACCAAAGCAGATTGCTAGGACTGGTACATG
# prediction: 1
# input sequence: TAACCGATTCTGAGCGCGCTGGTCAGTTAAGCATCATCGCGCGAGTTAGTCCAGGTACTTTACCGAGTAAGCGACCGTGAGTGAGGGTAAGTTGCCTTAA
# prediction: 0
# input sequence: TGACAGGTGCGCCTTTGATCTACGTAAAAAGGTGTCGAACATGTCCGCTAGAAGCGCCAGGCTAGACACTGTGTGTGACTTATACCGGGGTCTATGGTTA
# prediction: 1
# input sequence: TACGAGCGGCCCGATGAAAAGTAGCTCTGACAACCGTGCGCCGGGGGGCTACAACATCCTTGGTATAAGGTCTCGATGCCATGATGATCTATAGTTAATC
# prediction: 0
まとめと展望・ゲノム解析を越えて
前回のBlogで、HyenaDNAというLLMでDNAのシークエンスを分析するモデルを紹介しました。今回はその実践編として公開されているHyenaDNAのコードと事前学習済みのモデル・パラメータを利用した実行方法を簡単に紹介しました。このようにステップごとに見てみると、データはDNA配列とあまり馴染みないものではありますが、通常の機械学習のフレームワークとして扱えることがわかります。
さて、遺伝子の研究における1つのマイルストーンのあるヒトゲノム計画では、1990年当時に15ヵ年(2005年)を目標にヒトの全てのゲノム配列の解明するために開始されました。実際には技術の進歩と他のグループとの競争により計画は加速し、概要版が2000年に、完成版が2003年に発表されました。当時、ヒトゲノムの解析には1億ドルもの膨大なコストがかかっていましたが、次世代シークエンサー(NGS)の技術革新のために、現在では1,000ドル以下でまでコストが下がっています。(驚くべきことにムーアの法則を上回る脅威的なスピードでコストが下がっています。)
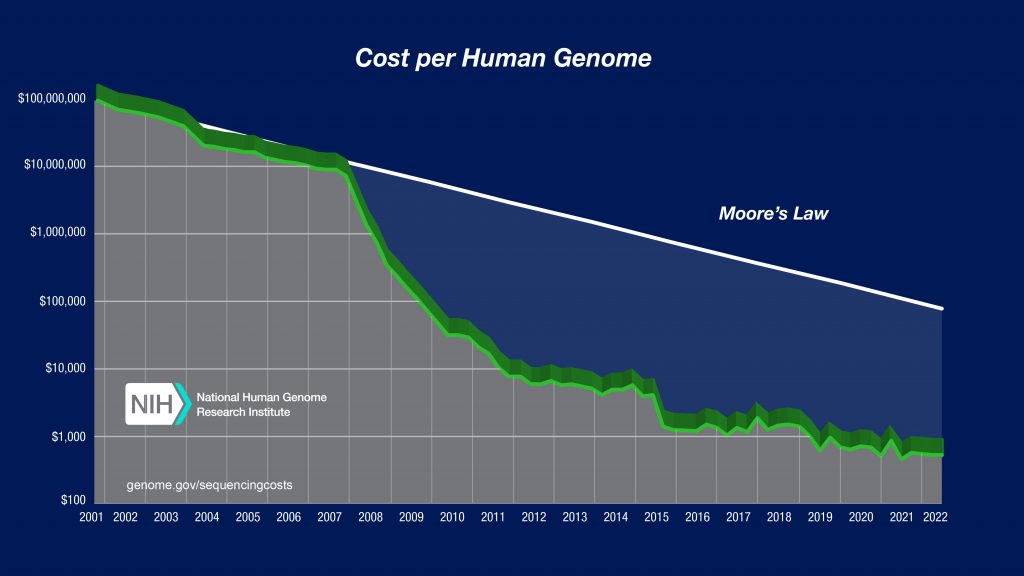
National Human Genome Research Institute The Cost of Sequencing a Human Genomeより引用
遺伝子配列の解析が安価かつ容易に行えるようになったことで、分析されるデータ量がこれまで以上に増えると期待されます。これは、遺伝子情報の利用を促進し、新たな研究や発見を生む可能性を秘めています。特に注目すべきは、Deep Learningの応用で、活発に研究が進められております(Awesome Deep Learning Single Cell Papers)。
しかし、ただの遺伝子情報を持っているだけでは、生命の全ての現象を説明することはできません。 我々の体は約37兆2000億個もの細胞(注: 身長172cm、体重70kgの30歳男性の推定値)からできていると言われています(はたらく細胞冒頭ナレーションより)。 これらの膨大な数の細胞は赤血球や白血球、肝細胞、筋細胞、神経細胞等、それぞれの機能に応じて分化しています。 遺伝子配列は共通ですが、各細胞で異なる遺伝子が複雑に作用し細胞の多様性を生み出しています(注;なお全細胞の3分の2を占める赤血球は成熟の過程で核やミトコンドリアなどの器官を失っています)。それらを解明するための細胞単位での遺伝子の転写物(Transcript)をその全て(Transcriptome)包括的に分析する研究領域はTranscriptomicsと呼ばれます。 さらには、全タンパク質を分析するProteomics、代謝物を分析するMetabolomicsなどの分野があります。 これらのオミクス(ome(すべての・完全) + ics(学問))と総称される研究領域は、それぞれに大量のデータが生成されることから、データサイエンスの活用が期待されています。Deep Learningなどの技術の応用により、生命現象をより深く理解することができると期待されています。
参考資料
- HyenaDNA: Long-Range Genomic Sequence Modeling at Single Nucleotide Resolution [https://arxiv.org/abs/2306.15794]
- hyena-dna [https://github.com/HazyResearch/hyena-dna]
- HyenaDNA: learning from DNA with 1 Million token context [https://hazyresearch.stanford.edu/blog/2023-06-29-hyena-dna]
- Genomic Benchmarks [https://github.com/ML-Bioinfo-CEITEC/genomic_benchmarks]
- OmicsML Awesome Deep Learning Single Cell Papers [https://github.com/OmicsML/awesome-deep-learning-single-cell-papers]
グループ研究開発本部の最新情報をTwitterで配信中です。ぜひフォローください。
Follow @GMO_RD