シングルセル解析入門:Covid19データセットで遊んでみた
こんにちは。AI研究開発室のK.S.(女性、外国人)です。
まず、宣伝させてください。久しぶりに、生物に関する記事を書きますが、少しでもこのブログ関連技術に興味がある方、是非、【新規プロジェクト】データサイエンティストに応募してください。
それから、本題です。今回のブログはシングルセル(Single-cell、単一細胞)解析の基本的な知識から簡単な解析(データの前処理)までを解説していきたいと思います。生物の専門家やバイオインフォマティクスの方は、シングルセルといえば、おわかりになるかと思いますが、エンジニアの方ですと、はてなが出てしまうかもしれませんので、シングルセルとは何か、何ができるか、から説明したいと思います。例として、知らない人がいない新型コロナウイルス(Covid19; COronaVirus Infectious Disease, emerged in 2019)のデータで遊んでみます。
目次
1. Single-cell(シングルセル)とは
シングルセルの説明前に、セル(細胞、Cell)を復習させてください。Cell「細胞」の語源はギリシャ語で、小さな部屋を意味します。細胞は全ての生物が持ち、体の中では一番小さいです。ヒトは60兆個(10-100兆個)の細胞があり、銀河にある星より多いです。それらの細胞は220種類(200-240種類)に分けられます。各種類はそれぞれの特徴を持ちます。また、細胞の中には色々なものが入っています。細胞を国とすると、国を管理している政府は核(nucleus)です。また、国内で様々な役割があり、エネルギー発電所はミトコンドリア(mitochondria)、焼却炉(不要物などのゴミ処理の場所)はリソソーム(lysosome)、タンパク質の合成場所はリボソーム(ribosome)です。ちなみに、遺伝子の情報はとても貴重ですから、核外への持ち出しは禁止です。そこで、DNAは似た構造のmRNA(messenger RNA)に情報を転写されます。それから、mRNAは核外に出てリボソームと結合し、tRNA(transfer RNA)が転写情報を元に必要なアミノ酸(タンパク質を構成している成分)をリボソームまで運んできます。リボソームの一部はrRNA(ribosome RNA)です。シングルセルは単一細胞で、シングルセル解析では、これらの小さな細胞を一つずつ観察して、それぞれが何をしているかや、どんな特徴を持っているかを調べます。細胞ごとに遺伝子と呼ばれる情報があり、それによって私たちの体がどのように機能するかが決まります。このシングルセル解析では、それらの情報を読み取り、その情報から体の仕組みや健康状態を理解しようとします。これにより、病気の原因を見つけたり、新しい薬を開発したりするのに役立つのです。
レビュー論文によると、シングルセル解析は様々なレベルで解析できます。主に、Transcriptome, Chromatin Accessibility, Surface Protein, Adaptive Immune Receptor Repertoire, Spatial Resolutionが挙げられます。
- Transcriptome(トランスクリプトーム):
トランスクリプトームは、ある生物学的なサンプル(例: 細胞、組織)が特定の時点で発現している全ての遺伝子のRNA産物(mRNA)の総称です。トランスクリプトーム解析は、遺伝子発現のパターンや量を理解するために重要な手法です。 - Chromatin Accessibility(クロマチンアクセシビリティ):
クロマチンはDNAとタンパク質からなる染色体の構成要素であり、アクセシビリティはクロマチンが転写因子や他の転写制御因子にどれだけアクセス可能かを示します。クロマチンアクセシビリティ解析は、遺伝子の発現調節やクロマチンの構造に対する洞察を提供します。 - Surface Protein(表面タンパク質):
細胞の外側に存在するタンパク質で、細胞の機能や特性に重要な役割を果たします。これらのタンパク質は細胞の特定のタイプや状態を示すのに利用されます。 - Adaptive Immune Receptor Repertoire(適応免疫受容体レパートリー):
適応免疫系において、抗体やT細胞受容体などの受容体が特定の抗原に対する多様な変異を持つことを指します。これは、免疫系が異なる病原体に適応する能力を提供します。 - Spatial Resolution(空間分解能):
空間的なスケールにおける解像度のことで、特定のデータや情報が与えられた空間内でどれだけ詳細に示されるかを表します。高い空間分解能は、微小な構造や位置関係を観察する能力を示します。
2. Single-cell RNA sequencing (scRNA-seq)解析
Single-cell RNA sequencing(scRNA-seq)は、一つ一つの細胞が持つmRNA(遺伝子の情報を持つ分子)の量を測定する技術です。生体組織サンプルを取り出して、細胞を一つずつ分離します。その後、各個々の細胞のmRNAが測定され、遺伝子ごとにどれだけのmRNAが存在するかを調べます。実験では、細胞をプレート上に分けるプレートベース(plate-based)の方法や、液滴ベース(droplet-based)の方法(マイクロ流体液滴を使って細胞を捕捉する)があります。プレートベースは比較的感度が高く、細胞辺りの遺伝子数を測定可能ですが、並列に測定できる細胞数が少なく、より技術的なノウハウと個別細胞の取り扱いのスキルが必要です。今回は液滴ベースの方法に焦点を当てています。
このscRNA-seq分野で広く利用されるプラットフォームは10x Genomics社が開発したもので、重要な手順は下記です。
- 細胞準備:細胞を取得し、適切な準備を行ってシングルセルの状態にします。通常、細胞が個々になるようにディスペンサーやピペットで分離します。
- GEM(Gel Bead-In-Emulsion)反応:10x Genomicsの独自の技術であるGEM反応を行います。これにより、barcode(バーコード)が付与されたgel beadsと細胞がemulsion(分散質・分散媒が共に液体である分散系溶液のこと)になります。
- cDNA(complementary DNA)合成とライブラリの作成:gel beads内で、細胞からのmRNAが逆転写され、cDNAが合成されます。この段階で、barcodeとUMI(Unique Molecular Identifier)が付与されたcDNAライブラリが生成されます。
- 次世代シーケンシング:ライブラリを次世代シーケンサーでシーケンスし、遺伝子発現データを取得します。シーケンス時には、barcodeとUMIが付与された情報が得られます。
- データ解析:シーケンスデータを解析し、個々の細胞ごとの遺伝子発現パターンを特定します。クラスタリングや次元削減などの手法を用いて、細胞のタイプや状態を識別します。

上記のプロトコルで、なぜ単一細胞と遺伝子を特定できるのか。
細胞と遺伝子を測定するため、cDNA合成のステップで、Barcode(バーコード)とUMI(Unique Molecular Identifier)は、scRNA-seqなどの遺伝子発現解析で重要な役割を果たす識別子です。
Barcodeは、異なる細胞や分子の識別に用いられる短いDNAまたはRNAの配列です。各細胞や分子に固有のbarcodeが割り当てられ、このbarcodeを通じて、シーケンシングデータの中でそれぞれの細胞や分子を識別することが可能となります。barcodeは、細胞ごとに異なるものや、同じ細胞内の異なる分子ごとに異なるものが使われます。
UMIは、ユニークな分子識別子であり、同じ分子が何度も逆転写される問題を解決するために導入されました。同じmRNA分子が複数回逆転写されると、その分子は複数のコピーとしてカウントされてしまいますが、UMIは各分子にユニークなIDを付与することで、同一分子の複数の複製を区別します。これにより、遺伝子発現レベルをより正確に評価することが可能になります。
BarcodeとUMIを組み合わせることで、scRNA-seqで各分子や細胞を正確に識別し、遺伝子発現パターンをより精緻に解析することができます。そして、参照ゲノムを使用して、細胞ごとの遺伝子のカウントマトリックスを生成する生データ処理パイプラインで行われます。この過程で細胞ごとの特定の遺伝子のカウントが集計され、解析の出発点となるデータが作られます。しかし、絶対定量になっていないので、異なった実験結果を比較するときに、注意が必要です。
また、scRNA-seqの進歩により、高品質かつ効率的な実行が可能になりました。ただし、データには低品質の細胞などによる系統的でランダムなノイズが含まれるため、場合によって前処理が重要です。前処理では、品質管理、正規化、データ修正、特徴選択などが行われ、生物学的な解釈を正確に捉えることが目指されています。
一つな大きな前処理技術はdoublet detectionです。データを分析する前に、その辺を理解した上で気をつけないといけなく、注意点として説明します。
3. Doublet Detection
3.1. Doublet Detectionとは
doublet(ダブレット)は、scRNA-seqやその他のシングルセル解析手法において、1つのセル(singlet)と見なされるべき場所で2つ(ダブレット)の異なる細胞が誤って一緒に扱われた状態を指します。つまり、ダブレットは2つの異なる細胞が重なってしまった状態であり、これにより、解析の際に誤ったデータが生成される可能性があります。実験測定の技術的な限界で、doublet formationといいます。ちなみに、2つ以上重なった場合はmultiplexといいます。ダブレットは、異なる細胞の特徴や遺伝子発現パターンが混ざってしまうため、正確な解析が難しくなります。これを避けるために、ダブレットの識別や除去が重要です。最近の研究では、ダブレットの識別や除去のためのアルゴリズムや手法が進化しており、正確なscRNA-seq解析のための取り組みが進んでいます。
doubletやmultiplexは様々なツールがあり、DoubletFinder(R package), Scrublet, Soloなどです。9手法で検討しているBenchmarking Computational Doublet-Detection Methods for Single-Cell RNA Sequencing Data論文があります。今回はニューラルネットワークを使用しているSoloを紹介します。Solo: Doublet Identification in Single-Cell RNA-Seq via Semi-Supervised Deep Learning論文を参考にしながら、説明します。また、SoloはVariational AutoEncoder(VAE、変分オートエンコーダ)を使っていますので、Soloを説明する前に、VAEを復習します。
3.2. Variational AutoEncoder(VAE)の復習
Variational Autoencoder(VAE)は、コンピュータがデータを効率的に学習し、新しいデータを生成するための仕組みです。例えば、絵や音楽を学んで新しい絵や音楽を作ることができます。VAEは、2つの重要な部分で構成されています。まず、encoder「エンコーダー」と呼ばれる部分があります。これは、データを少ない情報に圧縮して表現します。次に、decoder「デコーダー」という部分があり、この情報をもとに元のデータを再構築します。
特にVAEは、latent space(潜在空間)と呼ばれる特別な場所を使います。ここでは、データを表現するための規則やルールがあります。新しいデータを作るとき、この潜在空間のルールに従ってデータを生成します。これにより、新しい創造的なデータを作り出すことができるのです。
VAEは、絵や音楽、様々なデータを学んで、それらに基づいて新しいクリエイティブなものを作るコンピュータの魔法のようなものです。
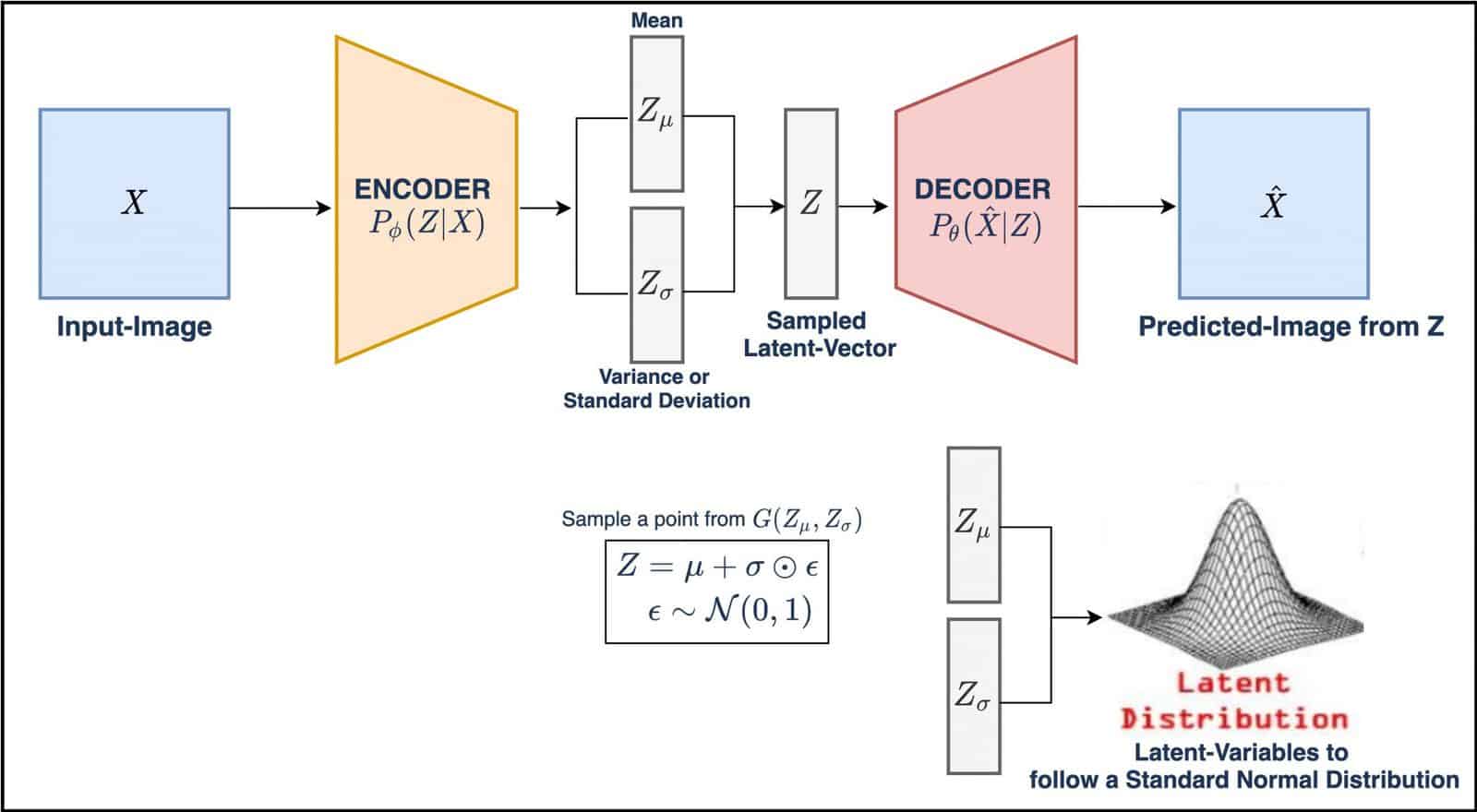
上記の図によると、近似関数\(P_{\phi }\left ( Z|X \right )\)は確率的エンコーダーであり、普通のオートエンコーダのエンコーダーと同様の役割を果たします。条件付き確率\( P_{\theta }\left ( \hat{X}|Z \right )\)は確率的デコーダーとしても知られる生成モデルを定義し、これはオートエンコーダのデコーダーに似ています。
VAEでは、二つのloss functions(損失関数)が使われていて、reconstruction lossとKL-divergence loss です。
- Reconstruction Loss:VAEは、入力データをエンコーダーして潜在空間にマッピングし、その後デコーダーして再構築することで訓練されます。Reconstruction lossは、元の入力データと再構築されたデータとの間の差を評価します。Reconstruction Lossは、通常、平均二乗誤差(Mean Squared Error, MSE)やベルヌーイ交差エントロピーなどの方法を使って計算されます。この損失関数は、入力データを効果的に再構築するために、ネットワークが潜在空間で意味のある表現を学ぶのに役立ちます。
- KL-divergence Loss:VAEは、潜在空間に分布を近似することで、新しいデータを生成します。通常、潜在空間の分布を正規分布(Normal distribution)と仮定します。KL-divergence Lossは、実際の潜在空間の分布と仮定した正規分布との間の差を計量します。この損失関数は、モデルが潜在空間で適切な分布を学ぶのを助け、潜在表現が適度に拡散していることを確保します。これにより、潜在空間が連続的で意味のある表現を持つようになります。
それでは、SoloはどうやってVAEに適応するのか話していきたいと思います。
3.3. Soloの紹介
上述のように、Soloはダブレットを測定するためのVAEベース手法です。計算によるダブレット検出法は、実験中のほとんどの細胞がシングルであると仮定し、観測されたデータからシミュレート(模擬実験)されたダブレットをin silicoで生成することで、ダブレット集団に対するおおよその見解を得ることができます。観測されたデータからシミュレートされたダブレットをin silicoで生成することで、ダブレット集団を近似的に見ることができます。3つの段階があります。1.doublet simulation(ダブレットのシミュレーション)、2.cell embedding (細胞の埋め込み)、3.classifier training(分類器の学習)です。

1. doublet simulation
観察された細胞をランダムに選んで合計し、擬似的な一重の細胞として扱うことで、ダブレットをシミュレートします。これにより、機械学習モデルを訓練して観察されたデータとこれらのシミュレートされたダブレットを区別する練習を行います。シミュレートされたダブレットの深さや数を、ベンチマークデータセットのテストセットの精度に基づいて、ダブレットの深さと数のデフォルトを選択します。$$ d_{n} = x_{1} + x_{2} $$
ここで、\(x_{1} と x_{2}\) は単一細胞トランスクリプトーム(count data)の実験データです。
2. cell embedding
遺伝子発現は低次元の多様体上に存在し、scRNA-seqの標準的な下流処理ではこれを利用して、行列因数分解法や非線形法により細胞を埋め込みます。これまでのダブレット同定法は、線形因数分解PCA(主成分分析)を適用して細胞を表現します。オートエンコーダーとして教師なしで訓練されたディープニューラルネットワークによって生成されるような非線形手法から大きな価値を実証しているようです。ここでは、Deep generative modeling for single-cell transcriptomics論文で提案したsingle-cell variational inference(scVI)法を適用してVAEを訓練し、観測データを再構成します。簡単に言うと、VAEはニューラルネットワーク変換q(z|x)を学習して細胞プロファイルxを潜在変数zにエンコードし、逆変換p(x|z)を学習して潜在変数を遺伝子発現空間にデコードすることによってパラメータ化された負の二項分布のもとで、元のカウントデータの尤度を最小化するように確率勾配降下法を用いて学習し、尤度を最小化します。ベンチマーク・データセットのパラメータースイープで、潜在空間のサイズをエンコーダとデコーダの隠れユニットの数とします。
3. classifier training
潜在空間にセルを埋め込んだ後、soloはscVIのニューラルネットワークの枠組みを継続し、標準的な識別的分類器をエンコードの最後に追加するようにしています。具体的には、観察されたデータに対して教師なしでscVIモデルを訓練します。次に、エンコーダを抽出し隠れ層を追加しダブレットの状態を予測します。scVIエンコーダのパラメータを凍結し、バイナリクロスペントロピーロスによる確率的勾配降下により、観測されたダブレットラベルとシミュレートされたダブレットラベルを予測するために、追加の分類器層を訓練します。ダブレットの分類中にscVI scANVI(single-cell ANnotation using Variational Inference)モデルを使用してエンコーダの重みを訓練し続けます。早期停止(early stopping)を使用した場合、オーバーフィッティングが発生し、汎化精度が低下します。いくつかのデータセットでは、単一の訓練エポックが平均精度を向上させます。平均精度は向上しますが、この訓練手順は脆弱であることがあるようです。VAEエンコーダには、モデルの不確実性を反映するためにセルを確率的に埋め込むという利点があります。その結果、エンコーダの分布からエンベッディングをサンプリングすることで、汎化精度を向上させる正則化が可能です。また、scVIモデルは細胞のシーケンシングの深さを反映するために、ライブラリサイズパラメータをエンコードします。この特徴量を分類器に渡すことが、効果的な性能を発揮するために重要だそうです。
それでは、雰囲気を捕まえるため、簡単にscRNAseqデータに触れてみたいと思います。
4. Covid19のscRNA-seqデータで遊んでみた
今回のブログはCovid19のscRNA-seqデータで遊んでみたいと思います。やりたいことはデータ取得と簡単なデータの前処理(doublet detection)です。4.1. 実装環境とデータ
実装環境はgoogle colaboratoryを利用し、追加で下記のsingle-cell解析関連libraryをインストールしました。!pip install scanpy !pip install scvi-tools !pip install scikit-misc
4.2. データ取得
データはA molecular single-cell lung atlas of lethal COVID-19論文が提供したデータ利用し、scRNA-seqデータはNCBIから、生データをダウンロードしました。# download raw data !wget 'https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSM5226574&format=file&file=GSM5226574%5FC51ctr%5Fraw%5Fcounts%2Ecsv%2Egz' -O GSM5226574_C51ctr_raw_counts.csv.gz !gzip -d GSM5226574_C51ctr_raw_counts.csv.gzScanpyを利用し、データを取得してみます。ScanpyはAnnDataをベースにして作成させるため、データ構造はAnnData classになります。最も基本的なレベルでは、AnnDataオブジェクト adataは、データ行列 adata.X、オブザベーションの注釈 adata.obsと変数 adata.varをpd.DataFrameとして、構造化されていない注釈 adata.unsをdictとして格納します。オブザベーションと変数の名前は、それぞれadata.obs_namesとadata.var_namesでアクセスできます。AnnDataオブジェクトは、データフレームのようにスライスすることができます。例えば、 adata_subset = adata[:, list_of_gene_names] とします。

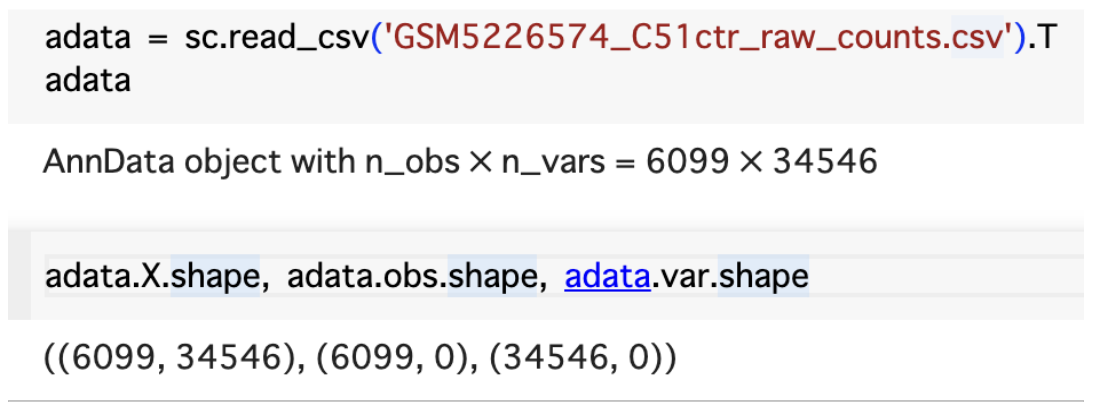
import pandas as pd import seaborn as sns import anndata as ad import scanpy as sc filename = 'GSM5226574_C51ctr_raw_counts.csv' adata = sc.read_csv(filename).T # csvファイルの場合 # adata = sc.read(filename) # h5adファイルの場合 adata取得結果は下記です。さまざまな遺伝子発現パターンが行列に含まれていることが分かります。
4.3. Doublet Detectionの実装
それでは、soloを使って、doubletを特定してみたいと思います。コードは下記ですimport scvi
sc.pp.filter_genes(adata, min_cells = 10)
sc.pp.highly_variable_genes(adata, n_top_genes = 2000, subset = True, flavor = 'seurat_v3')
scvi.model.SCVI.setup_anndata(adata)
vae = scvi.model.SCVI(adata)
vae.train()
solo = scvi.external.SOLO.from_scvi_model(vae)
solo.train()
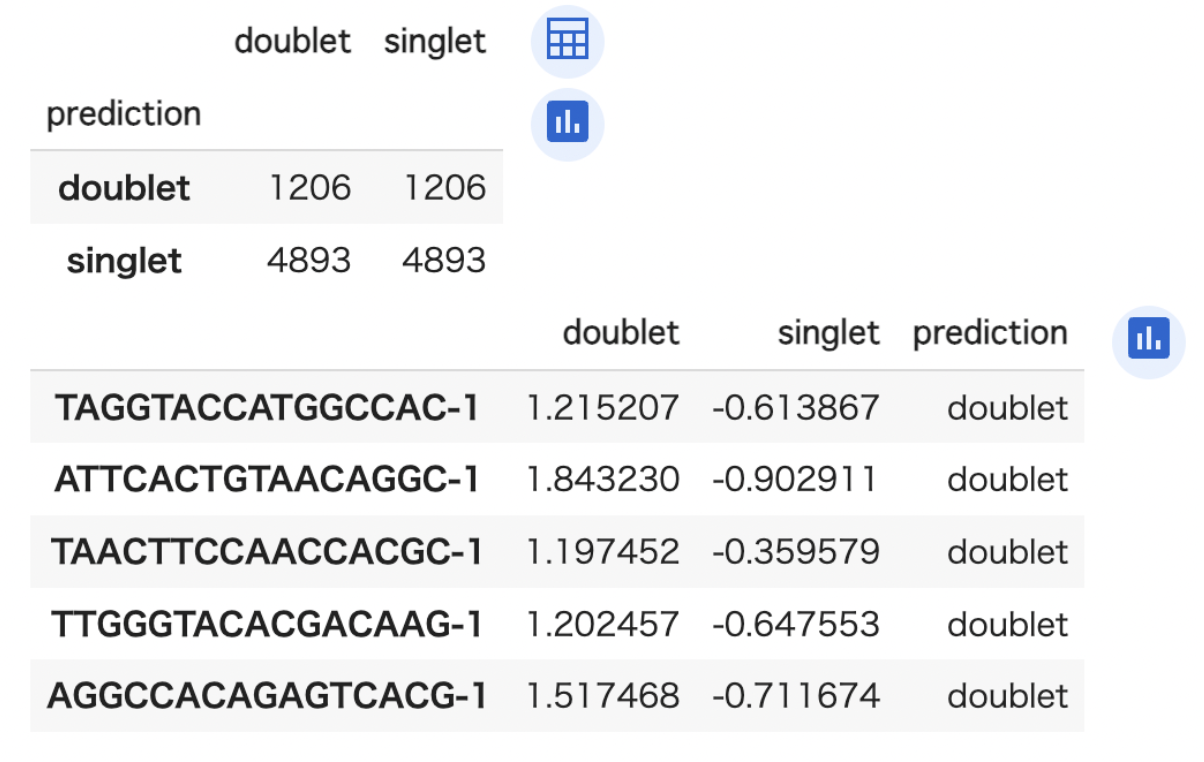
df = solo.predict()
df['prediction'] = solo.predict(soft = False)
df.index = df.index.map(lambda x: x[:-2])
display(df.groupby('prediction').count())
df.head()
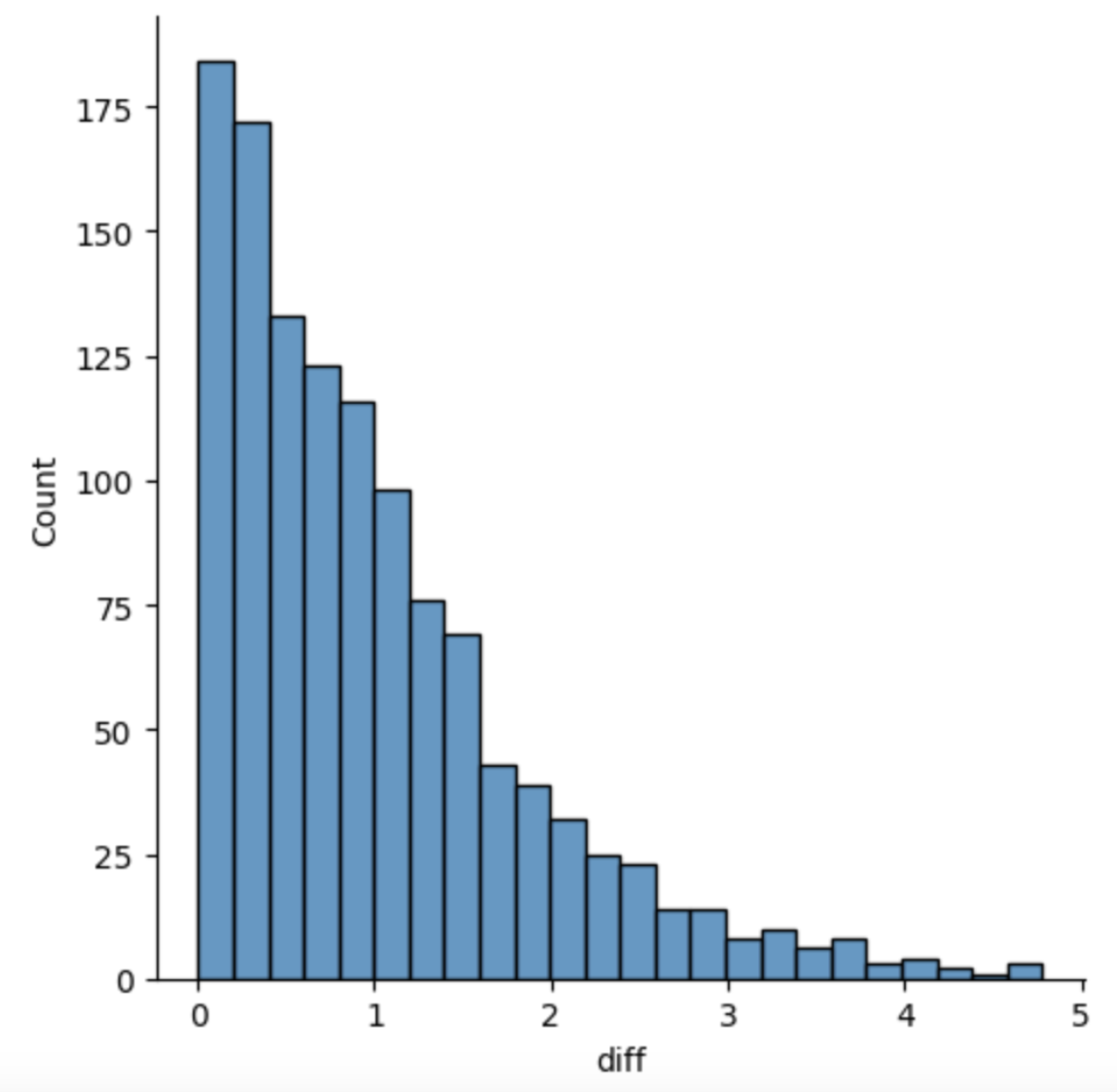
df['diff'] = df.doublet - df.singlet sns.displot(df[df.prediction == 'doublet'], x = 'diff')
5. まとめと考察
今回はscRNAseqとはなにかを説明し、簡単なデータの前処理を実装してみました。前処理だけでも、様々な技術が検討されており、解析のをはじめるまでにはまだ先が長いようです。最後に
グループ研究開発本部 AI研究開発室では、データサイエンティスト/機械学習エンジニアを募集しています。ビッグデータの解析業務などAI研究開発室にご興味を持って頂ける方がいらっしゃいましたら、ぜひ 募集職種一覧からご応募をお願いします。皆さんのご応募をお待ちしています。一緒に勉強しながら楽しく働きたい方のご応募をお待ちしております。